Lab 13 – Soil eDNA Metabarcoding Analysis

4/18/19
Objectives/Goals: The objective of this lab was to learn how to effectively perform metabarcoding analysis. We did this by using sample data from an eDNA sample extracted from soil associated with the rhizosphere of Bermuda grass from the Blackland Prairie Soil, and running the data through qiime2 to get the taxonomic qzv. files to visualize and analyze.
Procedure:
- Open terminal
- Activate qiime2 [source activate qiime2-2019.]
- Download box files
- Make the directory for the files [cd /Users/sophiashaikh/Documents/CILICURE_2018]
- Import the sequences as a qiime2 artifact [qiime tools import \–type EMPPairedEndSequences \–input-path emp-paired-end-sequences \–output-path emp-paired-end-sequences.qza]
- Demultiplex the sequences to align the data [qiime demux emp-paired \–m-barcodes-file sample-metadata.tsv \–m-barcodes-column BarcodeSequence \–i-seqs emp-paired-end-sequences.qza \–o-per-sample-sequences demux.qza \qiime demux summarize \–i-data demux.qza \–o-visualization demux.qzv]
- Denoise the data with DADA2 to get rid of low quality sequences and create a feature table with representative sequences [qiime dada2 denoise-paired \–i-demultiplexed-seqs demux.qza \–p-trunc-len-f 220 \–p-trunc-len-r 220 \–o-table table.qza \–o-representative-sequences rep-seqs.qza \–o-denoising-stats denoising-stats.qzaqiime feature-table summarize \–i-table table.qza \–o-visualization table.qzv \–m-sample-metadata-file sample-metadata.tsvqiime feature-table tabulate-seqs \–i-data rep-seqs.qza \–o-visualization rep-seqs.qzvqiime metadata tabulate \–m-input-file denoising-stats.qza \–o-visualization denoising-stats.qzv]
- Create a phylogenetic tree [qiime phylogeny align-to-tree-mafft-fasttree \–i-sequences rep-seqs.qza \–o-alignment aligned-rep-seqs.qza \–o-masked-alignment masked-aligned-rep-seqs.qza \–o-tree unrooted-tree.qza \–o-rooted-tree rooted-tree.qza]
- Get the taxonomic classification [qiime feature-classifier classify-sklearn –i-classifier silva-132-99-515-806-nb-classifier.qza –i-reads rep-seqs.qza –o-classification taxonomy.qzaqiime metadata tabulate \–m-input-file taxonomy.qza \–o-visualization taxonomy.qzvqiime taxa barplot \–i-table table.qza \–i-taxonomy taxonomy.qza \–m-metadata-file sample-metadata.tsv \–o-visualization taxa-bar-plots.qzv]
- Drag the taxanomic qzv. file into the qiime2 viewer and analyze the eDNA
Data:
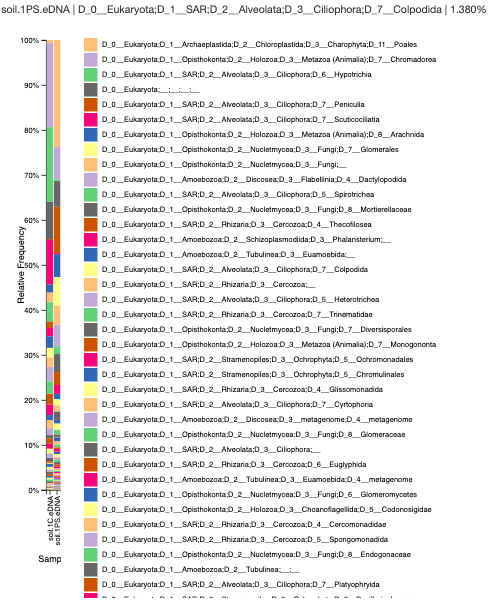
Analysis: The Chelex sample has a high amount of chromodorea DNA, a type of roundworm; the Powersoil sample, however, largely consists of the flowering plants, Poales. The Chelex seems to have a high abundance of nematodes and ciliates, but the Powersoil sample has a high abundance of plant material and amoebas.
Conclusion/Future Goals: It seems like I am getting the hang of using the terminal commands, and I can tell that I’m not struggling as much as I did the first lab we used the terminal/qiime2. I was able to look at the taxonomic bar graphs and determine the abundance and diversity of the sample. In the future, I would like to be able to use the common commands without having to refer to a guide at all. I would also like to use these processes with future research.