Lab 14: Poster Edits and Abstract 04/25/19

Purpose:
The purpose of this lab was to prepare for our poster presentations by making edits based on comments/critiques made. Another focus was to make a final draft for an abstract that will be submitted for the CURE symposium. We organized all the information (meta-data) from our samples in order to analyze our samples after they have been sequences.
Procedure:
- Vote on CILICURE logo, and what to do with it
- Organize metadata into excel sheet
- Make edits to poster and submit for printing, upload to box file
- Collaborate with team members to make a final abstract that will be submitted
Excel Metadata:
| section | group | label | GPS location | species | BHD | pH | Soil texture | Extraction | DNA concentration |
| 25 | 4 | GRS25_2Sp19 | -97.1182, 31.5498 | Quercus virginiana | 57cm | 6.4 | clay loam | silica bead | 262.1 |
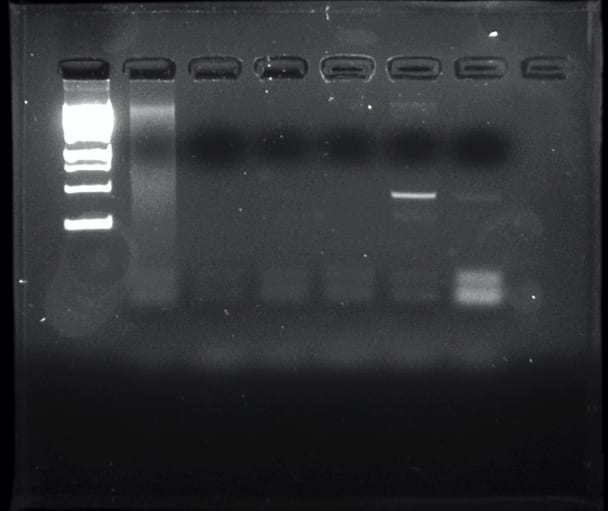
- The PCR result was also negative
Poster:
Abstract:
Title:
Soil eDNA Analysis Targeting the V4 Region Using the 18S Primer
Abstract
Soil ciliates are a diverse group of eukaryotes that play an important role in the environment but are understudied due to a lack of standard methods used to extract them from soil. The purpose of this experiment was to determine ciliate biodiversity in soil samples taken from the rhizosphere of Baylor trees. The protocols used were ones that have yet to be perfected in hopes of establishing them as universal method such as silica bead extraction of DNA and the chelex extraction method. DNA was extracted and purified from the soil collected using the ‘silica bead method’. The DNA concentration was obtained using a nanodrop and gel electrophoresis by running against a mass standard, and used PCR to amplify the 18s V4 region. The Polymerase Chain Reaction yielded a negative result, indicated by a ‘smear’ of DNA rather than a band. The nanodrop showed the concentration of DNA to be 262.1 ng/µl with the A260/280 to be 1.5. Despite negative results, the experiment showed the validity of results that can be found through silica bead and chelex DNA extraction; however, the lack of positive results was most likely due to an improper dilution or a degradation of DNA. Additionally, further research can be done using bioinformatics to visualize the ciliate diversity.
Word Count: 215
Discussion/Future Goals:
After working through edits for the poster I am becoming more excited to share our research with others. We will continue to practice presentations to the class next week before our presentation at the CURE symposium. Hopefully soon we send our DNA off for Illumina Sequencing and analyze it using QIIME2 and Cyverse. We will be able to use these programs in order to determine the biodiversity of the organisms, and hopefully ciliates, on our soil sample that was collected at the beginning of the semester.