Lab #14: Poster Presentation and Abstract Submission- 4/25/19

Megan Hudson April 25, 2019
BIO 1106-22
Lab #14: Poster Presentation and Abstract Submission
Section I: Objective
The purpose of this lab was to edit and submit our final abstract and poster for the CURES Symposium presentation. We also ensured that all metadata was recorded for the soil samples to allow Dr. Adair to pick the most viable sample to send off for sequencing.
Section II: Procedure
- Vote on CILI-CURE Logo for possible merchandise
- Record sample identifier, GPS location, tree species, BHD (cm), pH, soil texture, extraction method, DNA concentration, volume, PCR results (+ or -), and soil label on bag into Metadata File Spreadsheet
- Label DNA tube with AHM22_5Sp19, concentration, and volume of sample
- Place the tube with a new label into microfuge tube rack
- Take the remainder of the lab to edit poster and abstract within lab groups.
Section III: Results
Metadata File:
| Group Members: | Megan Hudson, Madison Ambrose, Kelsey Menzie |
| Section | 22 |
| Group | 5 |
| Soil ID | AHM22_5Sp19 |
| GPS Location | -97.1203258, 31.5467120 |
| Tree Species | Quercus virginiana |
| BHD (cm) | 188.4 |
| pH | 6.5 |
| Soil Texture | Sandy Clay |
| Extraction Method | Silica |
| DNA Concentration | 1.44 |
| Volume | 30 ul |
| PCR | – |
| Soil Label on Bag | KRM22S19 |
Abstract:
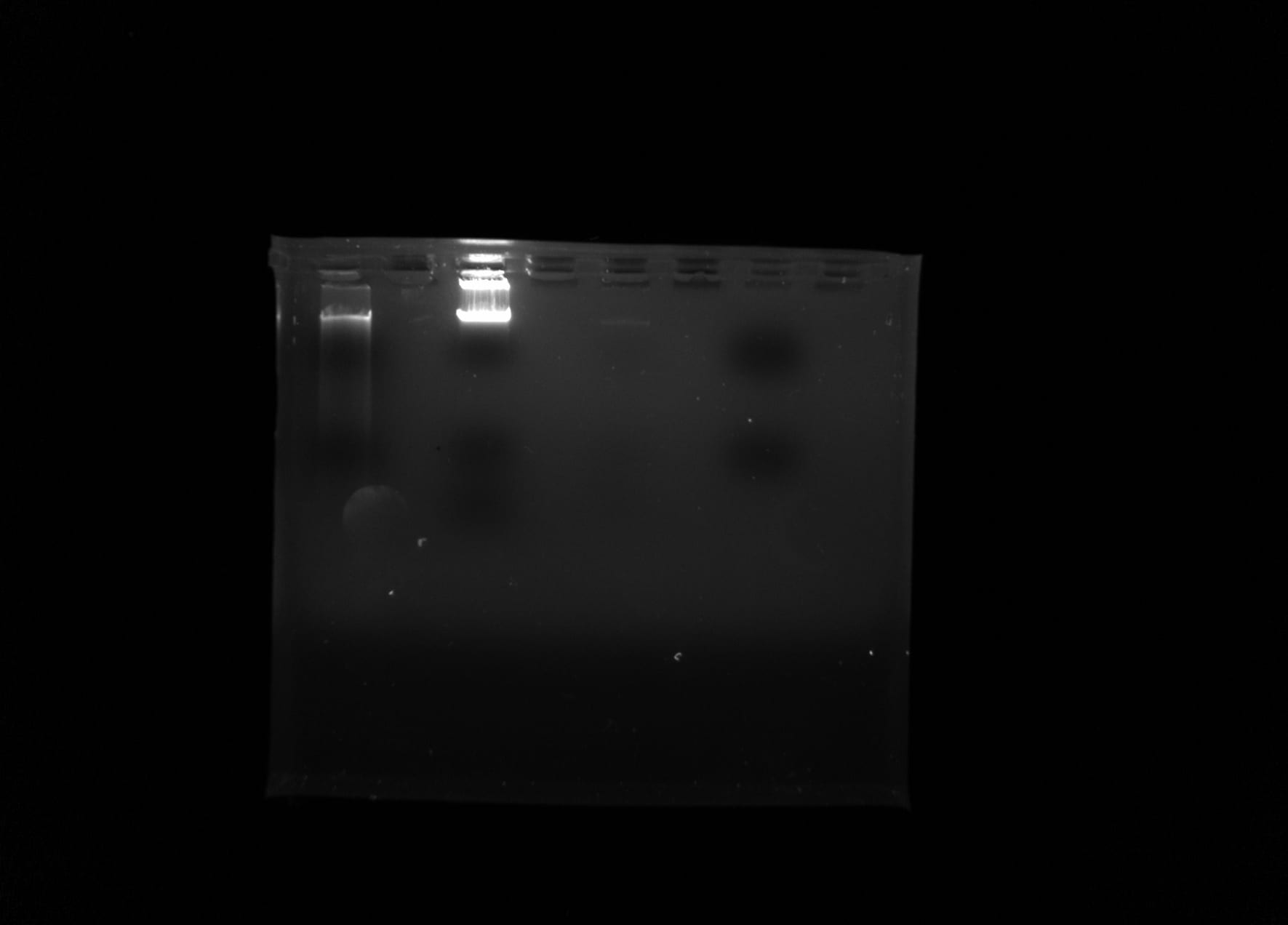
The biodiversity of microorganisms within terrestrial ecosystems is dependent on the viability of the community profile. Soil microbiomes and the influence of eukaryotes within is largely unexplored. The following field experiment analyzes soil collected from the rhizosphere of a Quercus virginiana to attempt to further classify ciliates and understand the diversity of one of the most controversial monophyletic groups, SAR. Metadata regarding soil texture, percent water content, and pH were obtained from each soil sample. The MNH soil sample was identified as sandy loam and KRM and MAA soil samples were identified as sandy clay. The samples were then isolated and eDNA was extracted using silica beads. The eDNA was then cleaned with 80% isopropanol alcohol and primed with an 18S V4 primer to isolate the V4 region of DNA in eukaryotes. The MNH and KRM samples underwent gel electrophoresis to attempt to view bands of DNA and KRM sample was amplified using a polymerase chain reaction (PCR). Results from the PCR amplification were analyzed using the NanoDrop 2000 Spectrophotometer and the MBC C305 UV gel imager. The MNH sample yielded an A260/280 concentration of 1.3 and the KRM sample yielded 1.44, indicating that DNA was present in the sample. Though it was shown that DNA was present, gel electrophoresis images produced no visible bands. These results indicate that soil collected from the rhizosphere surrounding the Quercus virginiana did not contain quantifiable eDNA for eukaryotes, despite ciliates being observed and cultured before soil eDNA was extracted.
Final Poster:
Section IV: Where your sample was stored
All soil sample bags were labeled at the beginning of the semester and stored under the fume hood and samples that produced positive PCR and gel electrophoresis results were put into deep freeze.
Section V: Conclusion and Discussion
Posters and abstracts were finalized and submitted so the poster could be printed, and the abstract could be uploaded to the brochure for CURES Symposium next week. Metadata was recorded in the class spreadsheet so samples that are viable enough to send off for sequencing have proper data for future studies. In both the KRM and MNH soil samples, results from the NanoDrop indicated a presence of eDNA, but when we ran our gels and performed PCR, results were negative and did not show a presence of DNA. Since our results were negative for eDNA abundance, most likely our samples will not be sent for sequencing. Although my groups’ samples did not produce positive results, we still were able to establish a methodology for DNA extraction and PCR amplification for future CILI-CURE labs. Moving forward, my group plans to practice presenting our poster so we are prepared for CURES next Friday. As the semester comes to an end, I am grateful for all the research experience and scientific writing skills I have learned in CILI-CURE.