April
25
Lab 14 – Final Poster and Abstact

4/25/19
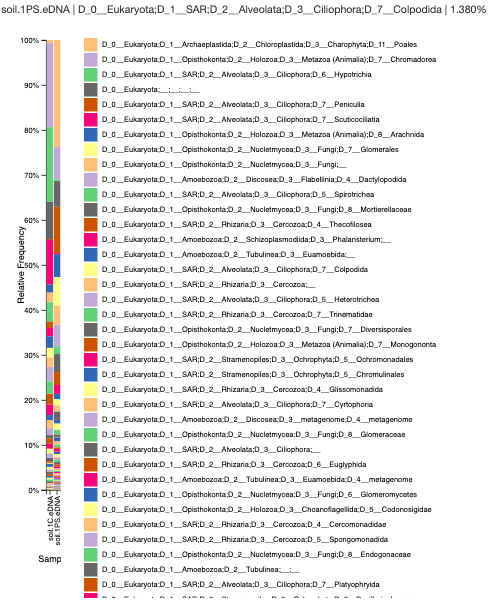
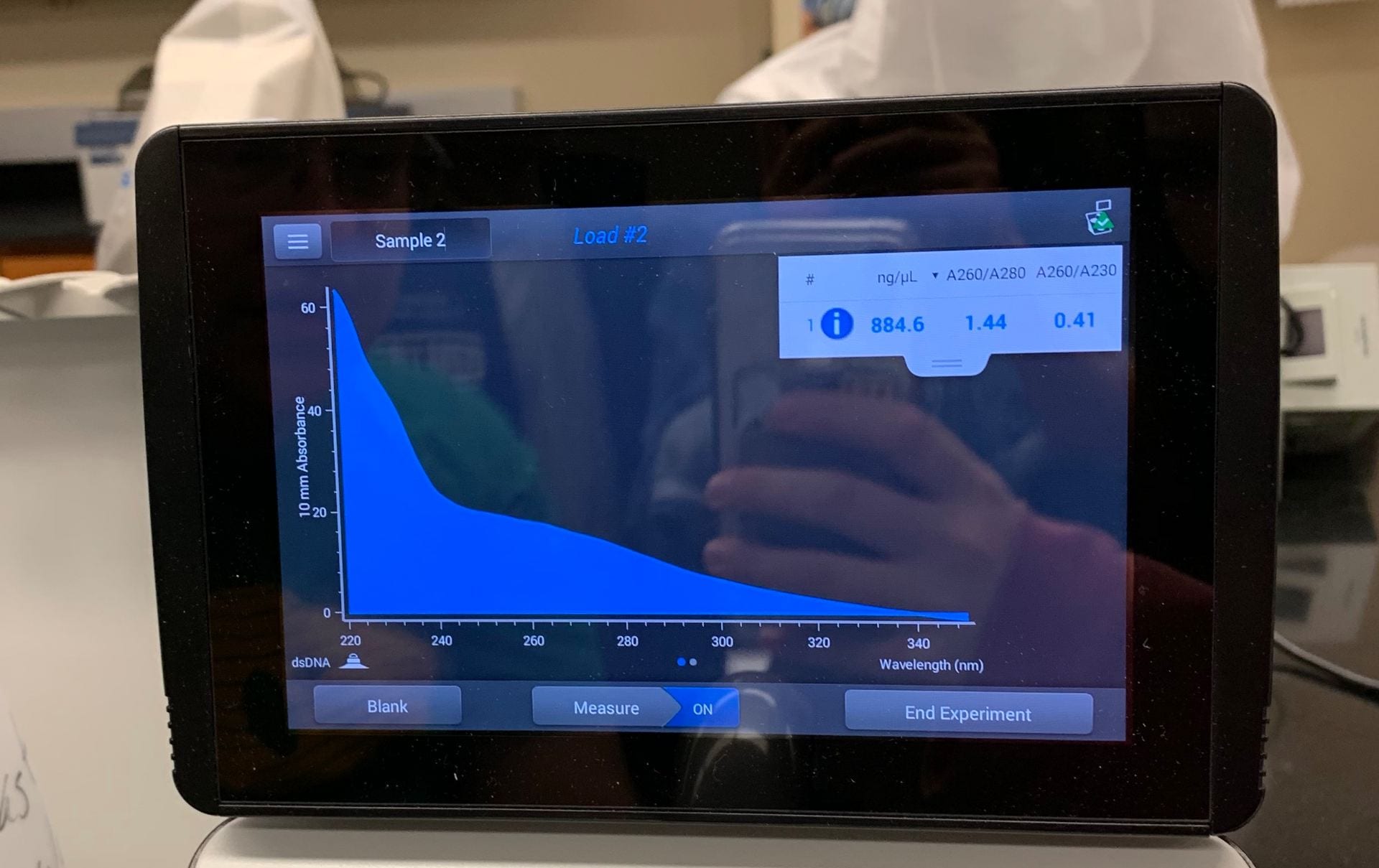
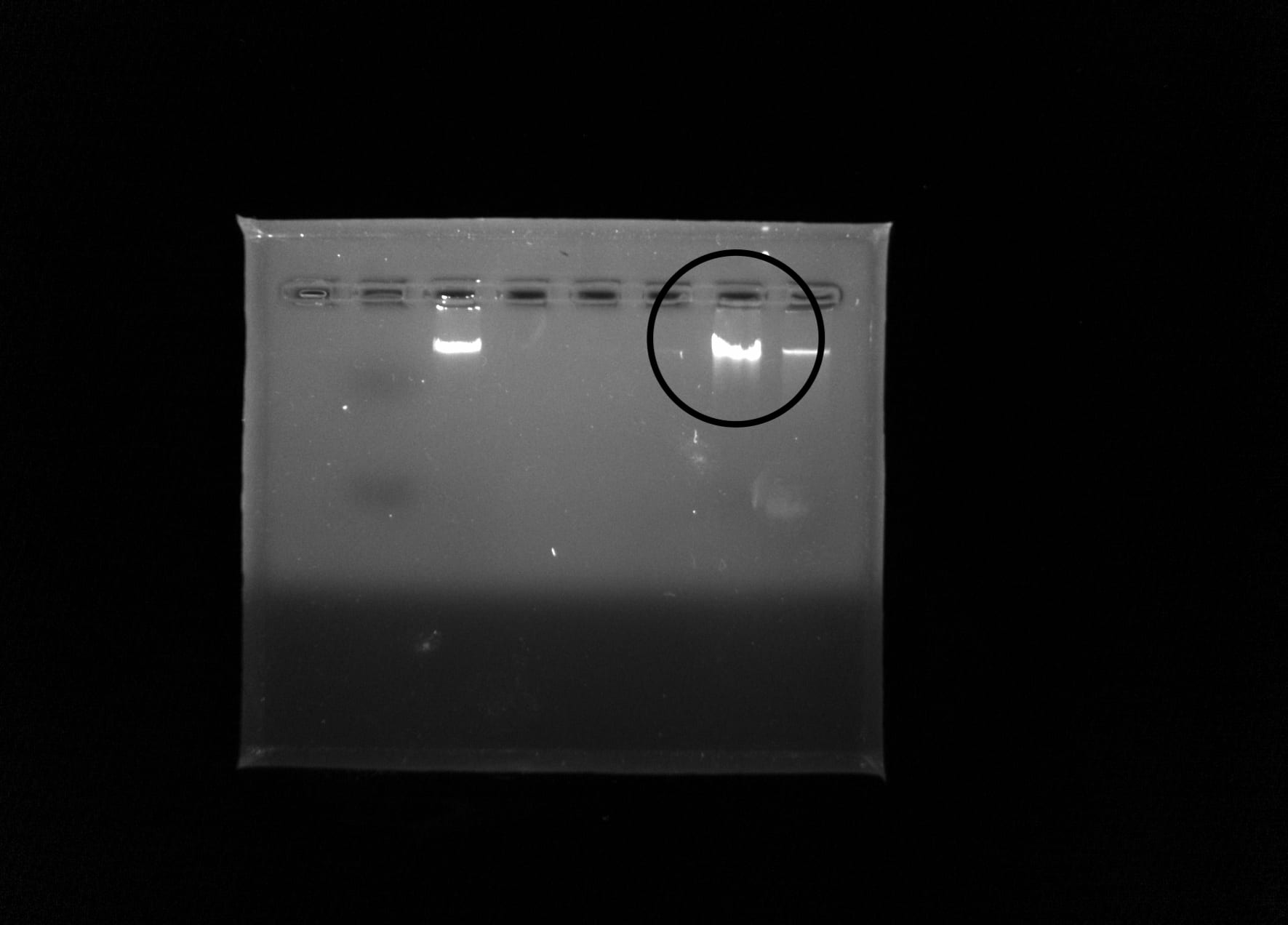
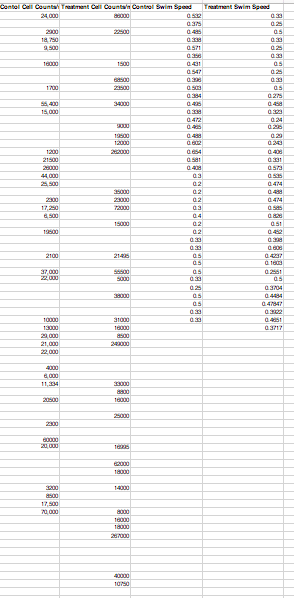
Objectives/Goals: The objective of this lab was to tie all of the loose ends by voting on a logo, creating an abstract, finalizing the posters, and uploading our metadata to a class excel file.
Procedure:
- Vote on lab logo
- Upload metadata to class excel file
- Write and submit abstract to box files
- Fix poster according to Dr. Adair’s canvas comments
- Upload final poster to box files
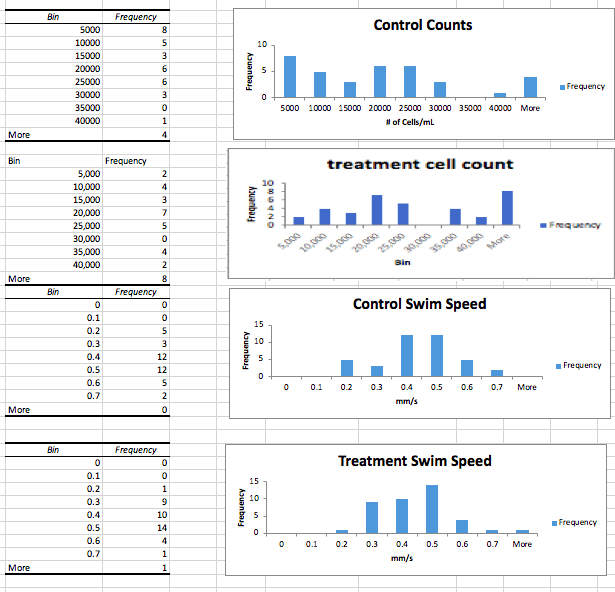
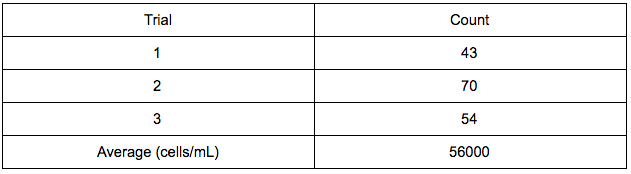
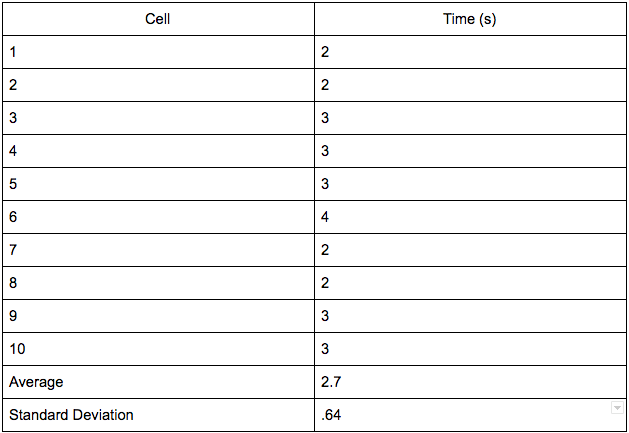
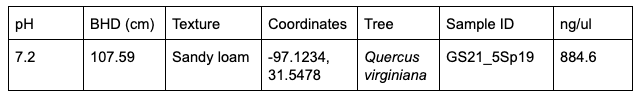
Data:
Conclusions/Future Goals: Overall this lab went smoothly because it was mostly doing touch ups on the poster and uploading files to the Box. Our poster has all of the necessary data, and our abstract accurately and briefly summarizes our experiment. In the future, I hope that I can conduct an experiment and efficiently make a scientific poster to communicate my findings and why they are important.