Objective:
In lab our goal is to learn how to quantify DNA using the “nanodrop” technique, how to set up and run a PCR reaction as well as learn to make an agarose gel. We hope to successfully complete the PCR reaction after learning the procedure, and achieve the end goal of being able to complete the metabarcoding process and be able to analyze as well as classify soil ciliates.
Purpose:
The purpose of today’s lab is to continue the PCR process and testing positive and negative controls as well as the DNA extracted from the soil sample. The purpose of learning how to make a gel and actually physically creating it, is to be able to successfully run gel electrophoresis. After reading the scenific literature provided from our pre-lab, we are able to conceptualize how the cox1 gene primers bind to the DNA and how this will allow us continue in our metabarcoding process.
Procedure:
*Use bleach to create a sterile work station. This will help avoid any contamination from other microbes and bacteria on the lab tables.
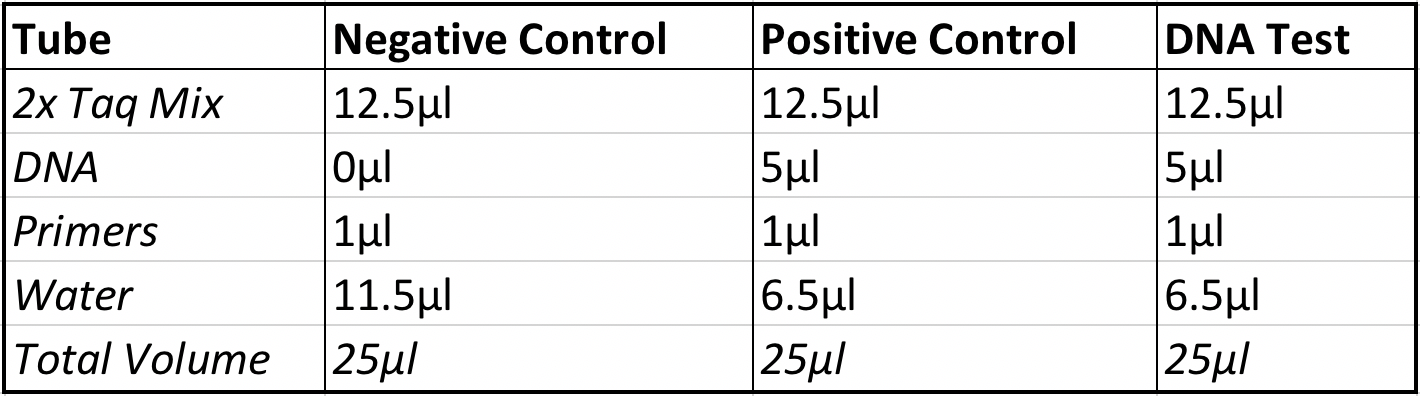
1.) Obtain three small tubes containing 12.5 micro-liters of the TAQ polymerase mixture.
2.) Label the tubes, “DNA test”, “positive control”,and “negative control”.
3.) Add 5 micro-liters of the DNA template sample to the tubes labeled “DNA test”, and “positive control”.
4.) Add 1 microliter of the cox1 primer to each of the three small test tubes.
5.) Add 11.5 micro-liters of H2O to the negative control tube.
6.) Add 6.5 micro-liters of water to the DNA test tube and the test tube labeled positive control.
7.) Add up the volume of everything added into the tubes to ensure that each has a final volume of 25 micro-liters.
8.) Place the tubes in a tube rack at the front of the room, record the place of each of the rubes within the rack.
9.) Store the tubes at 4 degrees Celsius or freeze until confirmation through gel electrophoresis may be performed.
10.) Proceed to making the gel for gel electrophoresis by adding 0.6 grams of agarose to 40mL of 1 x TAQ in an erlenmeyer flask. (Weigh the agar on a scale using weigh paper, tare the balance after placing the weigh paper on the scale and add agar powder until there is 0.6 grams).
11.) Cover the top of the flask with weigh paper and then place the lid loosely over both to prevent it from exploding in the microwave.
12.) Place in the microwave at power 7 for 1 minute and 20 seconds, or until boiling.
13.) After, place in heat bath for about 5 minutes then add ethidium bromide.
14.) Pour the mixture in a gel mold and label is with your group identifier (LSK G#5).
15.) Let sit for 30 minutes then store in the refrigerator for next lab.
Data/ Observations:

COX 1 Gene Forward Primer:
5′-ATGTGAGTTGATTTTATAGAGCAGA-3′
COX 1Gene Reverse Primer:
5′-GGDATACCRTTCATTTT-3′
PCR Cycling Conditions for COX 1 Primers:
Stage 1: 94 degrees Celsius for 4 minutes.
Stage 2: 5 cycles of denaturation at 94 degrees Celsius for 30 seconds, annealing at 45 degrees Celsius for 1 minute and extension at 72 degrees Celsius for 105 seconds.
Stage 3: 35 cycles of denaturation at 94 degrees Celsius, annealing at 55 degrees Celsius for 1 minute and extension at 72 degrees Celsius for 105 seconds.
Stage 4: Hold at 72 degrees Celsius for 10 minutes.
*We were required to where gloves for sterilization purposes and because ethidium bromide is a carcinogen.
*After pouring the gel into the mold, we believe that the liquid did not distribute equally before it started to harder, causing it to have a sense dense area of the agarose mixture around the wells.
Storage:
The three small tubes with the DNA test, positive and negative controls, were placed under the projector in a tube rack labeled on one axis with numbers and the other axis with letters. The DNA test tube is located in slot B1 labeled “DNA” on the side and a “5” that represent group number five. The positive control tube is located in slot B2 and is labeled with a “+” on the side and a “5” on the top and the negative control tube is located in slot B3 and is labeled with a “-” on the side and a “5” on the top. After creating a gel, we let it sot for thirty minutes so it could take the shape of the mold. It was later left on the end of our lab tables to be picked up and stored in the refrigerator so that we may use it during the next lab. The gel mold has a piece of blue tape that is labeled “LSK G#5” which stands for “Lindsey, Sandi, Kaitlyn Group # 5” which we have created as our group identifier.
Future Goals:
After completing the steps of PCR and creating a gel, I hope that we are able to run gel electrophoresis and are able to view the DNA fragments under the UV light. Our gel was not evenly distributed throughout the mold; I hope that this will not effect the DNA fragment traveling through it. I am somewhat worried that we will not be able to clearly see and DNA markers on the ladder in the gel because of our soil samples. In the future, I would like to be able to continue on with the metabarcoding process and hope the see the DNA fragments with in the gel so we can compare it to the markers of other organisms and classify them.