2-8-2018
Purpose: The purpose of today’s lab was to introduce us to a new way of pipetting using serological pipettes. Using the serological pipettes gave us some more confidence in that were accurately measuring our samples. We also tried a new protocol during lab today (third time’s the charm!!).
Procedure:
- Weight and tare the weighing dish
- Collect 5g of fresh top soil and while screening it from debris, place it in the weigh boat. After collecting, place it in a plastic vial.
- Using a serological pipette, add 10mL of water to the vial and mix for 5-10 minutes. The vortex can be used for this part.
- After mixing, allow the soil mixture to sit for 1-2 minutes to allow the sand to settle at the bottom.

- Use a micropipette to transfer 3.68 ml of the soil water into a new clean glass tube.
- Allow Mike to place 368 µl of 25% Glutaraldehyde into our vial containing our sample.
- Close the cap on the vial and mix with a vortex for 30 seconds to a minute
- After vortexing, inject 4 mL of sample into the previously Ludox filled tubes on our tables 2 mL beneath the top.
- Next, place 2 ml of the colored water on top of the Ludox solution layer.
- Weigh the vial and ensure the opposite group at your table have the same weight. If the weight is off, add more colored water to the vial with the lesser mass.
- With a sharpie, label the tube. (LAK on the sides and a star on the top)
- Centrifuge the Ludox solution for 15 minutes in a swinging bucket rotor at 4300 g`s.

13. Using a p1000, remove a total of 4ml of the cell layer.
14. Place 2 ml of the cell layer in one tube and another 2ml in another tube.
15. Label these 2 tubes also. (LAK on the sides and a star on the top).
This is all we were able to complete in lab today, but the rest of the protocol is listed below and will hopefully take place today during open lab.
- Centrifuge the 2 ml tubes again at 3000xg for 5 minutes to pellet the ciliate cells.
- Using a micropipette carefully remove the supernatant.
- Dispose of the waste liquid in the correct waste container.
- Place 100 uL of the PBS buffer to each pellet in their respective tubes.
- Resuspend the pellet by flicking the tube up and down.
- Place both tubes of cell suspension pellets from the two 2ml tubes into a single tube for a total of 200 uL
- Place five 2 uL drop s from the created 200 µl solution on a concave slide and count the cells using a 40X lens. Record the number of cell/uL then obtain a class average.
- If necessary add iodine in a 1:1 mixture. This can be done by placing 10 uL of stain with 10 uL of cells in a microfuge tube to be mixed and counted for five 4 uL drops. Each drop will have 2 uL of cells.
Control sample procedures:
The same procedure will be followed for the control as the original experiment, but instead of using the soil, we will obtain 5g of autoclave soil. The control sample will be our positive control in this experiment because we know there are ciliates in it.
Clean up Procedures:
- Place serological pipettes back into their sleeve and into the waste box.
- Any soil remains should be placed into a bucket as to not get any soil in the sink.
- Place any Glutaraldehyde into the glass waste bottle and rinse the tube with bleach and water.
- The Ludox tubes should be placed in the rack by the sink.
- Wipe the tables down with Windex.
Results/Observations:
The mass of our soil alone was 5.0g.
The mass of the tube containing the soil, water, Glutaraldehyde and Ludox solution was 41.3 g.
The mass after pipetting 10 ml of water in to soil was 14.0g.
The mass of our two 2ml tubes containing the cells after centrifugation was 3.5 g.
Storage:
All of the tubes used in my group were labeled with LAK on the side and a star on the top. The 2 tubes that contains the 2 ml of the soil, water, Glutaraldehyde mixture is located on the green rack. The conical tube was placed in the waste rack by the sink.


Error: We had an oopsy today in lab. Our calculations were wrong and we accidently added 12% more Glutaraldehyde that what we needed. The correct amount should have been 320 uL instead of 368 uL. This may result in a hard time counting cells because they might be unable to come out of their cyst form.
Conclusion: This was trial run 2 from the protocol we did last week. Some changes we made were the amount of glutaraldehyde and the amount of starting Ludox. Hopefully these changes help us see our cell layer better and as a whole works far more accurate and efficient. I’m excited to go to open lab to see the results. Even though we made a mistake in calculations of glutaraldehyde and this is our third protocol, I really think this could be the perfect one, and it’d be awesome for future labs to know we created this.
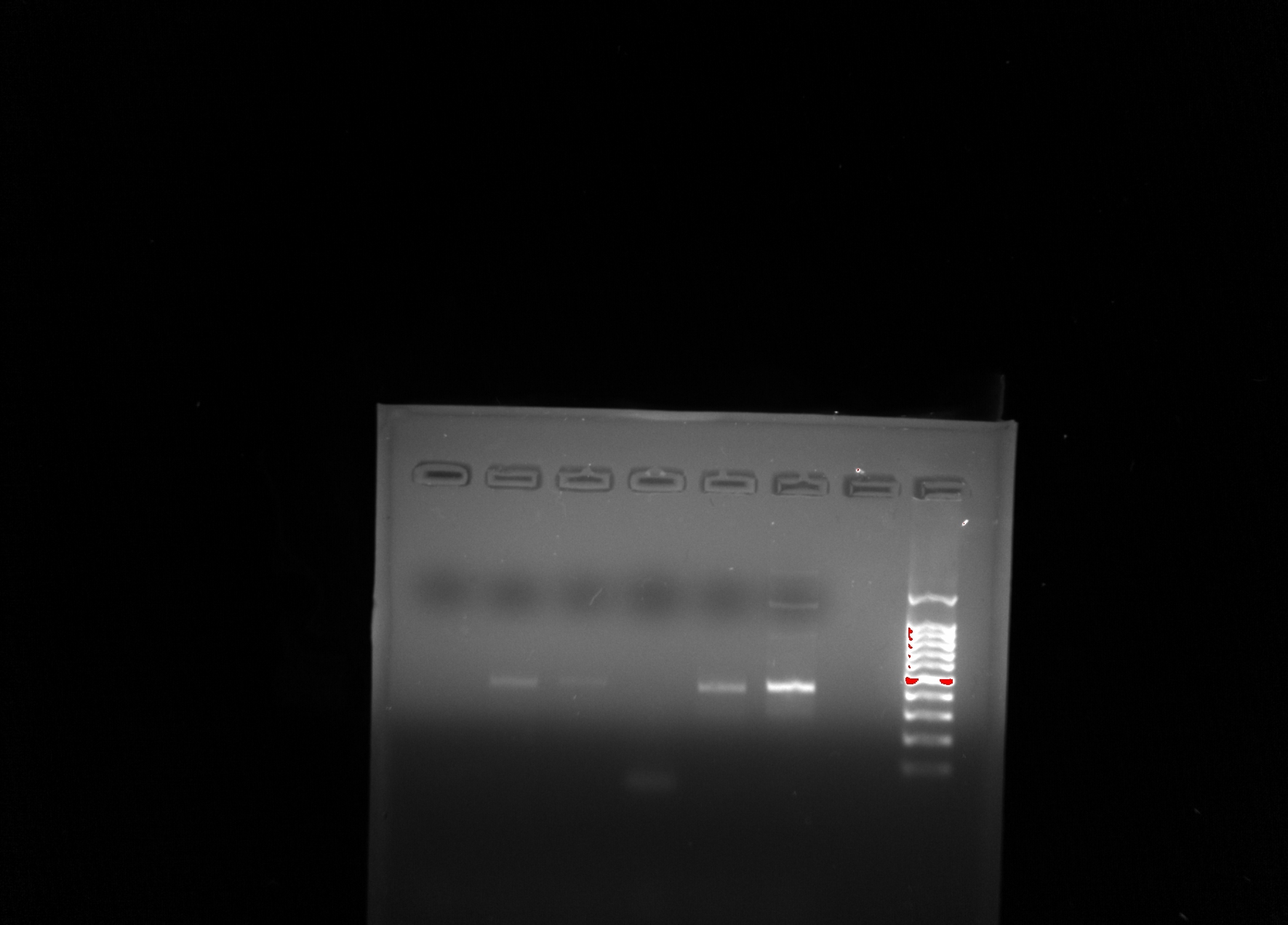
Future studies: For next lab, I hope to start looking at the DNA of the cells and continue onto PCR and gel electrophoresis. I actually did an experiment in high school with electrophoresis and bacteria on table tops, bathroom stalls, and office pens, so I can’t wait!