Lab 14: Poster Workshop 4/19/18

Purpose:
To allow students to revise their abstract and work on their poster presentation.
Procedure:
- Have a group discussion about the information that goes on each section
- Write down what goes on each section
- Revise what you wrote
- Make a poster presentation on powerpoint.
Data:

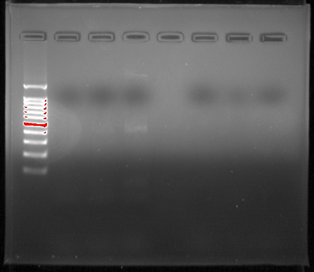
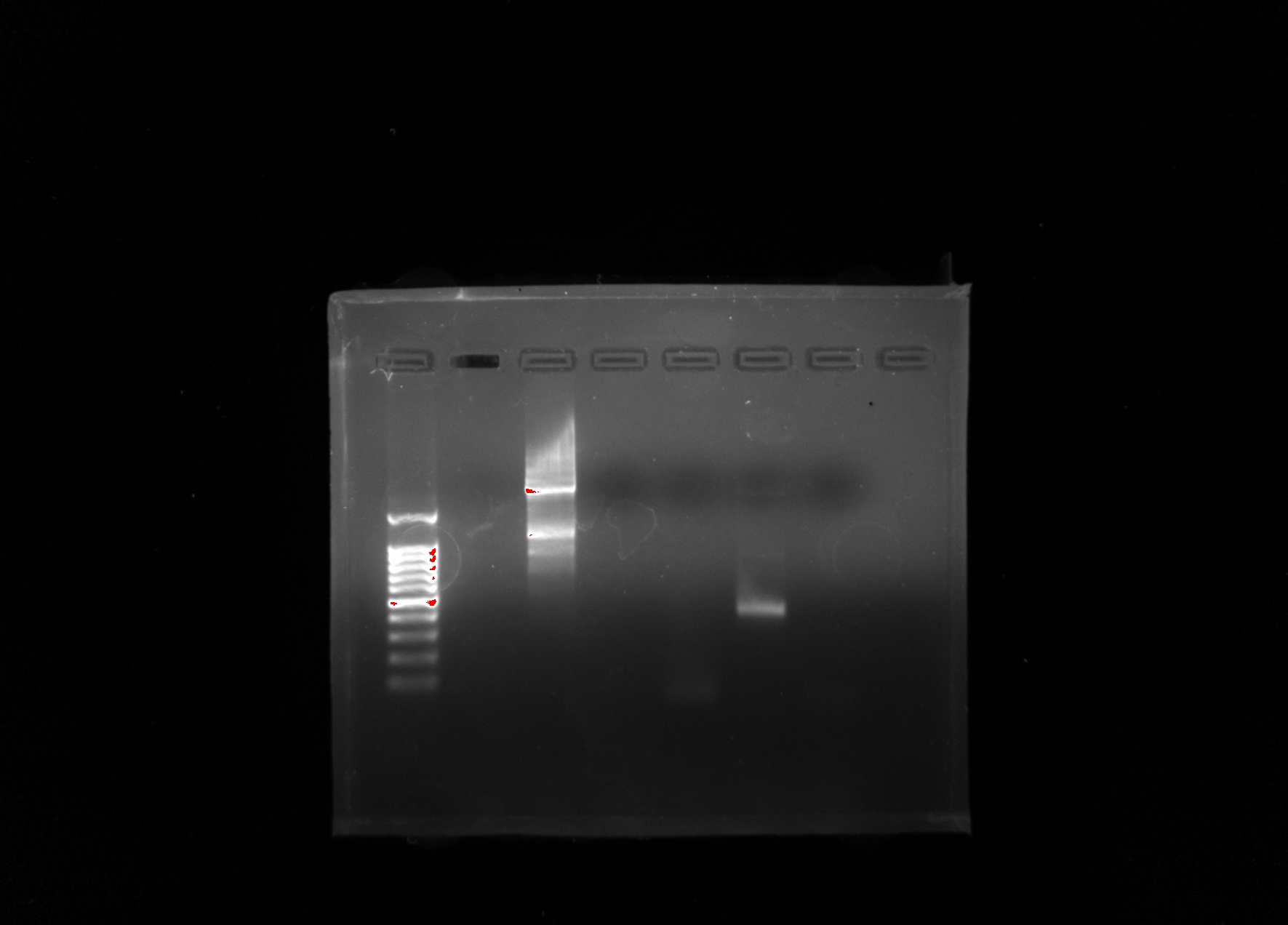
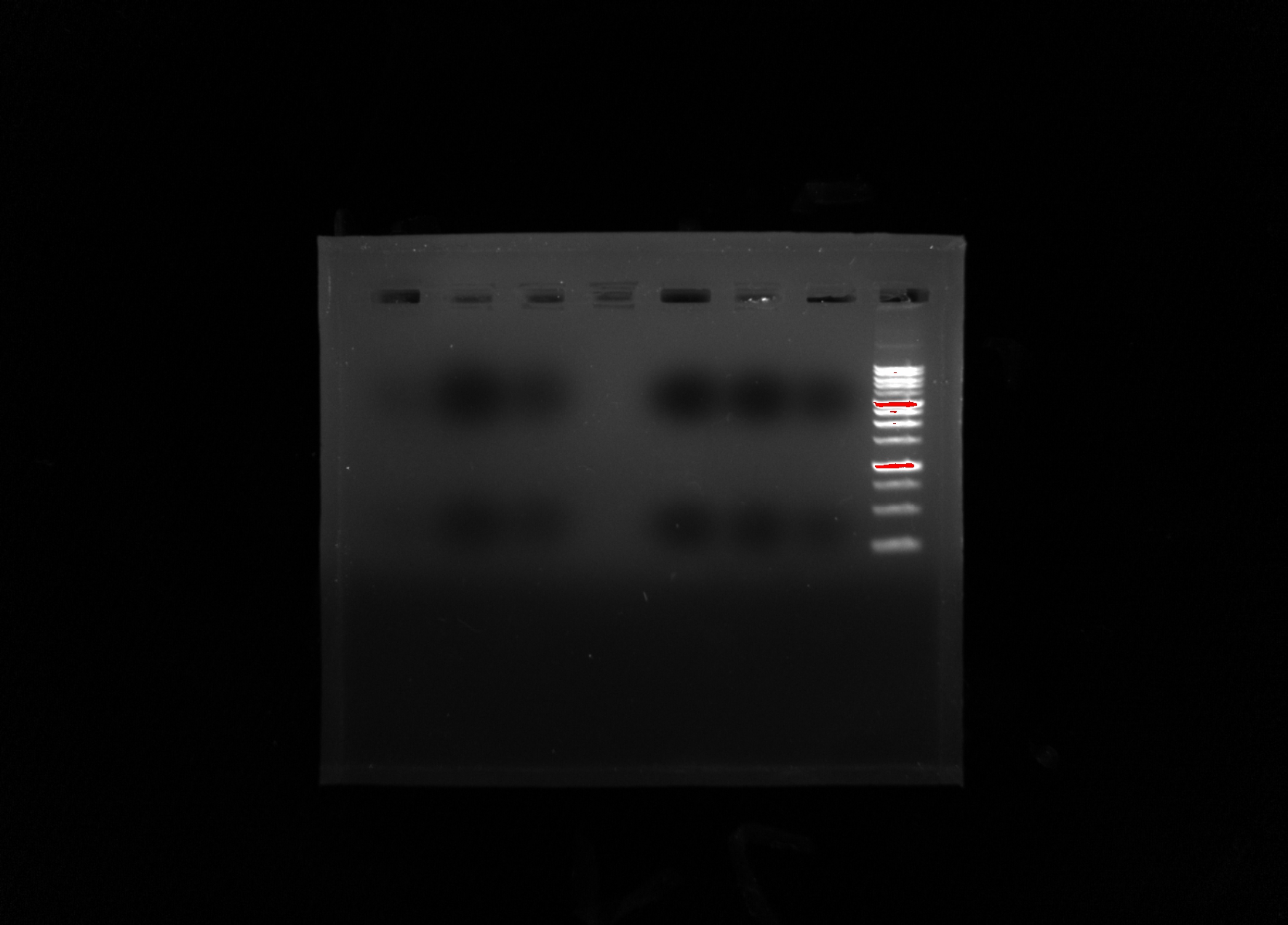
My group’s poster is shown above.
Abstract:
Organisms within the soil are important because they are what help sustain all life on Earth. Scientists have begun to recognize that there is a large diversity of soil protists whose existences have not yet been known, simply due to the lack of knowledge in the scientific community about soil biodiversity. In order to provide a solution, a correct protocol needs to be found to extract the DNA found in samples and what primers should be used to amplify it. Multi-methodological approaches were used to link morphological, molecular, and functional information of distinct ciliates. Various protocols were used: LudoxGradient, E.Z.N.A, Gel Electrophoresis, Chelex, and Powersoil. The Chelexand the Powersoilprotocols showed the most promising results in comparison, so the Chelexand Powersoilmethods were the two that were concentrated on. Each protocol was followed meticulously, and the COX 1 and V4 primers were the focus of each PCR amplification. Some errors in precise pipette techniques and accurate measuring could have been apparent after taking into account the results of each protocol. A gel from the Electrophoresis Protocol was made each time in order to analyze the negative, positive, and the environmental DNA samples to determine if DNA was extracted. The Ludoxprotocol did not produce any positive results from the gel. The PowerSoil technique did not produce many positive results from the gel as well. The chelex protocol had the best results for positive results in the gel. For further experiments, multiple tests of a singular protocol will be conducted, in order to ensure maximum accuracy.
Conclusion:
In conclusion, my group finished a good working draft of the poster. The next steps are to revise the poster and practice presenting the poster.