Lab 2: Genome Annotation
Ashton Peckinpah
1/17/2017
Objective: Today we familiarize ourselves with the tools we will be using for the remainder of this semester. One being DNAMaster used to auto-annotate the genes and read the results of the annotated genomes.
Methods: After watching a few DNA Mastering guided videos, we walked through the process with the phage Amigo.
- We were first taught how to download DNA Mastering onto our own individual laptops.
- We used the DNA Mastering annotation guide to walk us through the process of annotating a genome.
- We then began setting up our preferences on DNAMaster:
-Click on the File menu within DNAMaster and select Preferences
-Click on Local Settings tab and Colors sub-tab. We changed the colors to help us decipher and identify differing RNA’s.
-Click on Codons sub-tab and make sure “Use TTG start codons” is checked.
-Template code SSC: CP: SD: SCS: Gap: Blast: LO: ST: F: FS: ST
-May change SD scoring matrix to Kibler 7 and the spacing weight matrix to Kerlin medium
-Click the Apply button
- Used phagesdb.com to download the FASTA file of the phage amigo.
- Then we opened the new FASTA file amigo and auto annotated it.
Auto-Annotation:
- Find Amigo file and click Open.
- Go to Export button and click “create Sequence from this entry only”
- From the “Extracted from Amigo.fasta” window, go to the Genome menu and select Annotation, Auto-Annotate.
- Click Annotate.
- Once your genome is annotated, click on the Features tab at the top of the genome’s window.
- Right click the “Name” row and expand the row.
- Click ORF’s button.
- Click on an ORF.
- Click on the button labeled RBS.
In conclusion: After completing the lab, we learned how to properly use DNA Master. We begin to understand the information we are looking at especially after having this good foundation for to start with.
Lab 3: Link BLAST
Ashton Peckinpah
Tuesday, January 24, 2017
Objective: Understand how to complete a BLAST, which can be considered a critical element of a gene annotation. The BLAST tool finds regions of similarity between the sequences and generates a report to show the comparison.
Methods:
- Download the Link fasta file from phagesbd.org.
- Open DNA master.
- Open file in DNA master.
- Copy and Paste the product of the amino acids of the gene you want into NCBI protein blast.
- Add the following annotation notes in preferences: SSC: CP: SD: SCS: Gap: Blast: LO: ST: F: FS:
- BLAST
-Copy and paste the code in the product window.
-Go to the protein blast option on NCBI and PhagesDB.
-Paste the code there.
-Run the gene.
-View results.
Results: there is a list that follows the colorful graph. The list shows the most and least similar genes
When learning what the annotation template codes stand for, refer back to the DNA Mastering Annotation Guide.
-SSC: start/stop codons
-CP: codeine potential
-SD: Shine Dalgarno Score (also includes z value)
-SCS: whether or not the start codon choice agrees with glimmer and gene mark
-Gap: the gap or overlap with the gene before it
-Blast: Blast on NCBI, HHPred, phages DB
-LO: longest ORF
-ST: starterator
-F: Function
-FS: Function Source
In conclusion:
We now understand how to use GeneMark to indicate if a gene is entirely covered as well as the coding potential following the gene. For me, I better understood how to annotate a genome after blasting it. Knowing that when an individual blasts, they are simply comparing one genome to another.
As a class, we annotated genes 7 and 8. The GeneMark potential covered the entire genome, the starterator agreed with the start site, and the genes showed the best SD score as well as the longest ORF.
I feel prepared to annotate genes in the future after what knowledge was gained through this lab.
Lab 4: Phamerator
Ashton Peckinpah
Tuesday, January 31, 2017
Objective: Understand how to use Phamerator correctly as well as incorporate the useful technology into a genome annotation.
Methods:
Phamerator:
- Open the Virtual Box application.
- Click on the Phamerator option.
- Click on the phages tab to the left.
- Search link and then click map (6 genomes when comparing)
- Note the Pham number, members, and cluster.
- Go back to the phages tab and select several other AN Arthrobactors.
- Once you have clicked on phages you need, Click map.
Annotations:
-Annotate assigned gene.
-BLAST (NCBI and PhagesDB)
-Observe conserved domains of NCBI
-Observe Phamerator results.
-Function of the genome?
*Understanding how to read a phamerator report is a very important part of this lab
In conclusion:
Phamerator compares the genes using multiple alignments. Overall, Phamerator helps to clarify our genes function by comparing it with other phams (usually similar).
Lab 5: Starterator
Ashton Peckinpah
Tuesday, February 7, 2017
Objective: Learn how to operate Starterator, this piece of technology may help clarify our start codon on our genes. It is helpful in an entire genome annotation.
Methods:
Starterator:
- Open the Virtual Box.
- Hit start and open Phamerator.
- Click on the phages tab and search for the phage of your choice.
- Click map.
- Note the Pham number, members, and cluster.
- Compare the recommended start site (indicated with blue) to that of Glimmer and GeneMark.
- Indicate if you agree with start recommended by Starterator.
- There will be a graph followed by a text summary
How to read Starterator:
- each track represents an ORF
- Each colored line represents a possible start codon
- the white lines represent gaps.
- The report lists the tracks and the names of the genes and suggested starts Results: for gene 7 Lore_draft
In conclusion:
Starterator is one last tool used to accurately identify correct start codons. This tool is really helpful and I know it will be helpful next week with my lab partner and I.
Lab 6: Annotating Lore Genes 23 & 25
Ashton Peckinpah
Tuesday, February 14th, 2017
Objectives: Beginning to work on the gene Lore today after weeks of preparation. I plan on completing annotations for gene 23 as well as 25 today.
Tools used and/or Methods: Glimmer, GeneMark, HHPred, NCBI, PhagesDB, Phamerator, Starterator
Results for Gene 23: Start: 14113bp, Stop: 14781 bp FWD GAP: 4bp Overlap
GeneMark confirmed that all potential, typical as well as atypical, is covered.
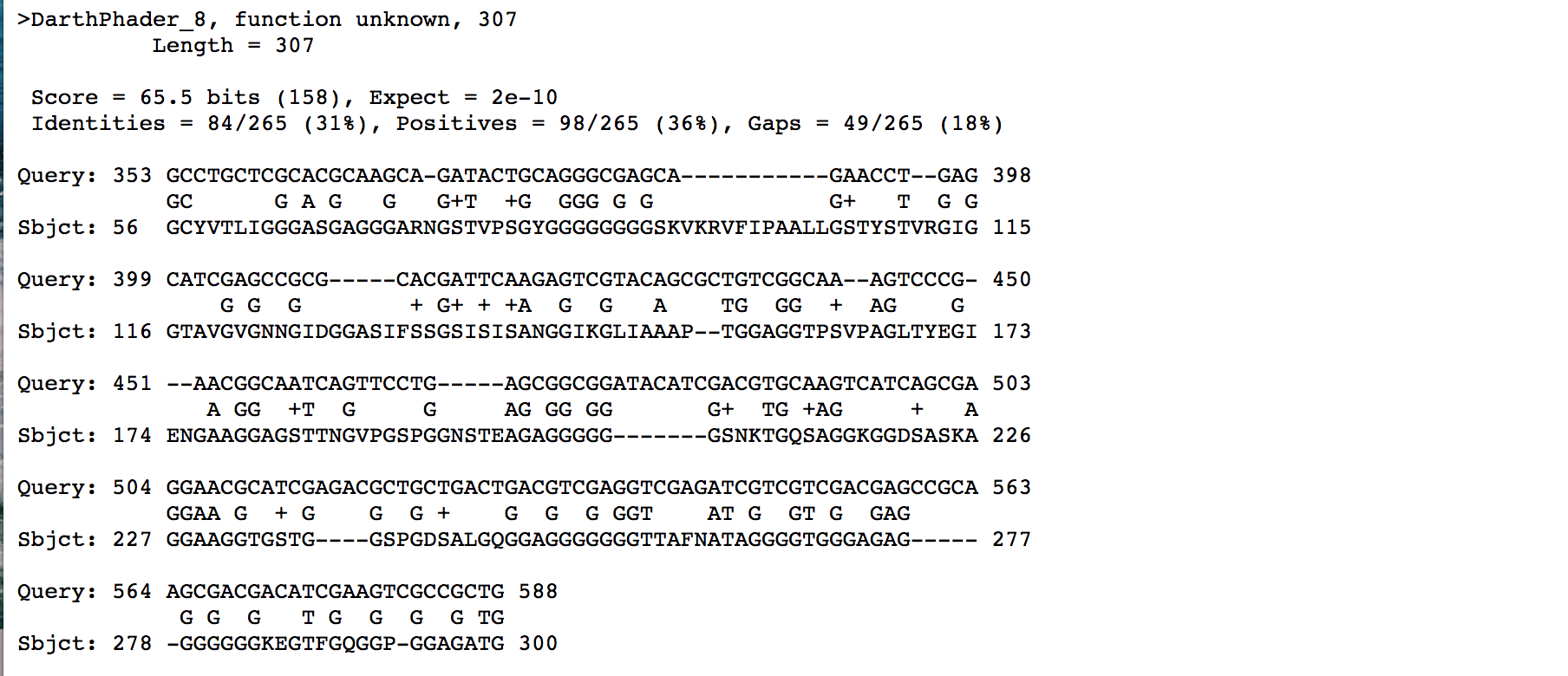
NCBI Blast identified an HTH superfam. Confirming it supports HTH DNA binding function, q:1 s:1
Phages DB blast: no known function for the gene
HHPred: supporter of HTH DNA binding function
Final Annotation Result:
Start: 14113bp Stop: 14781bp FWD GAP: 4bp Overlap SD Final Value: SD Score: -3.246 (Best score) Z-Value: 2.847 CP: The gene is covered SCS: Agrees with Glimmer, Disagrees with GeneMark Genemark called a reverse gene, but there was more potential in the forward gene that was not called NCBI BLAST: Helix-turn-helix DNA binding domain protein [Arthrobacter phage Decurro] q:1 s:1 E-Value: 2e-156 CDD: pfam13730, Helix-turn-helix domain E-Value: 1.09e-06 PhagesDB BLAST: StewieGriff_Draft_23, function unknown, q:1 s:1 E-Value: 1e-134 HHPred: Transcriptional Regulator E-Value: 5.9E-08 LO: Yes ST: Agrees with Starterator F: helix-turn-helix DNA binding domain FS: NCBI, HHPred, Phamerator Notes:
Results for Gene 25: Start: 14980bp Stop: 15276bp FWD GAP: 4bp Overlap
GeneMark confirmed that all potential, typical as well as atypical, is covered.
NCBI Blast: no known function for gene
Phages DB Blast: no known function
HHPred: no known function. Too high of an e value for the top hit.
Results for Gene 25:
Start: 14980bp Stop: 15276bp FWD GAP: 4bp Overlap SD Final Value: SD Score: -3.554 (Best score) Z-Value: 2.798 CP: The gene is covered SCS: Agrees with Glimmer, Agrees with GeneMark NCBI BLAST: hypothetical protein STRATUS_25 [Arthrobacter phage Stratus], q:1, s:1 E-Value: 4e-46 CDD: No good hit PhagesDB BLAST: StewieGriff_Draft_25, function unknown, q:1 s:1 E-Value: 2e-51 HHPred: No good hit LO: No All coding potential was covered with start position 14980 and LO had lower final score ST: Agrees with Starterator F: NKF FS: NCBI, Phages DB, HHPred Notes:
Lab 7: LORE Presentations
Ashton Peckinpah
Tuesday, February 21, 2017
Objective: Today we presented the genes we annotated so others may see and critic them as well. We also see what an entire genome looks like. As a class, we deciphered if a start codon change was necessary or if declared functions were correct.
Methods:
- Groups presented PowerPoint presentations to class.
- For others in the class not presenting, fill out the function.
-Was it auto-annotated?
-Were there any changes made to Lore?
- You fill out the Questions That Matter worksheet with the above information.
- We listened intently to others presentations. It helped me understand the annotation process listening to others experiences doing it.
In Conclusion:
This lab taught us how to successfully annotate an entire genome. Afterwards we were happy with our accomplishment of completing our first phage. It is exciting to think about these getting double-checked and then getting sent off to be published.
We begin annotating our next phage next week.